
Von Kleinwuchs spricht man in der Medizin wenn die Körperhöhe unterhalb der 3. Perzentile der Norm liegt, so dass definitionsgemäß 3% der Bevölkerung kleinwüchsig sind. Je weiter ein Mensch jedoch von der „Normgröße“ abweicht, desto problematischer wird sein Alltag. Dies betrifft einerseits das bauliche Umfeld, das auf Menschen einer gewissen Größe ausgelegt ist, und andererseits die Akzeptanz durch die Mitmenschen.
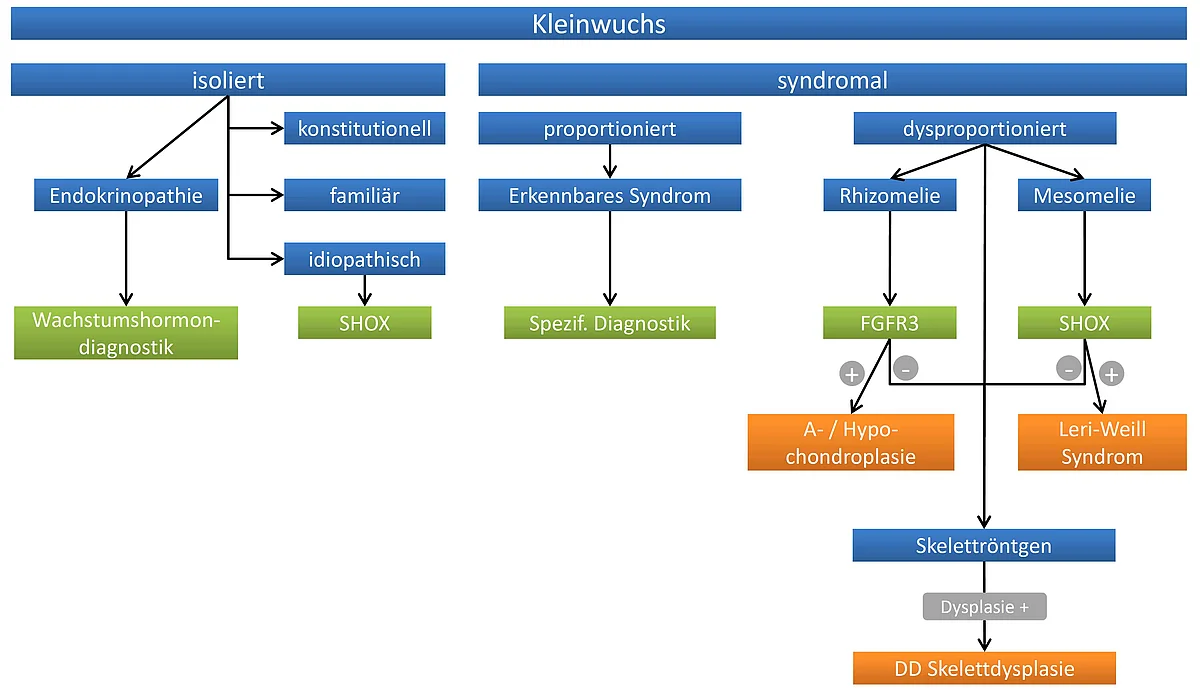
Klinisch unterscheidet man syndromale Formen, bei denen neben dem Kleinwuchs weitere Auffälligkeiten bestehen, von den nicht-syndromalen Formen, bei denen die Wachstumsstörung als isoliertes Problem auftritt. Daneben kann die Wachstumsverminderung mit einer Verschiebung der normalen Körperproportionen einhergehen (sogenannter dysproportionierter Kleinwuchs) und entweder nach der Geburt oder bereits intrauterin beginnen (sogenannter primordialer Kleinwuchs).
Ein dysproportinierter Kleinwuchs weist in der Regel auf eine gestörte Knochenreifung bzw. –differenzierung hin. Man unterscheidet derzeit 372 verschiedene Formen von solchen sogenannten Skelettdysplasien, von denen 215 auf Defekte in 140 bisher bekannten Genen zurückgeführt werden können. Hierbei konnten als ursächlich Defekte in extrazellulären Strukturproteinen, Makromolekülen, metabolischen Pathways, Signaltransduktions-Mechanismen, Kernproteinen, Oncogenen und RNA und DNA prozessierenden Genen nachgewiesen werden. Skelettdysplasien sind aber selten und treten insgesamt nur bei 0,04% der Neugeborenen auf. Daneben ist Kleinwuchs auch ein Merkmal zahlreicher seltener, komplexer Syndrome mit allgemeiner Entwicklungsstörung und von Störungen des Wachstumshormon-Signalweges. Die Fehlfunktion von Signalwegen, welche das Zellwachstum regulieren, können auch Erkrankungen wie Krebs, Diabetes mellitus und Infektionen zur Folge haben.
Abgesehen von den hormonellen Ursachen sind als Ursache für den idiopathischen Kleinwuchs bisher nur Defekte im Transkriptionsfaktor SHOX bekannt, welcher durch ein Gen in der pseudoautosomalen Region in Xp22.3 kodiert wird und bei ca. 2,4% der Patienten deletiert oder mutiert gefunden wird. Somit bleibt derzeit die Ursache des Kleinwuchses bei der Mehrzahl der Patienten ungeklärt. Die Aufklärung der molekularen Grundlage genetisch bedingter Erkrankungen ist jedoch nicht nur wichtig für Diagnostik, Prognostik und genetische Beratung der Betroffenen und deren Familien, sondern stellt auch die Voraussetzung für die zukünftige Entwicklung therapeutischer Interventionen dar.

