
Die Gruppe der Kurzrippen-Polydaktylie Syndrome (Short-rib polydactyly syndromes, SRPS) umfasst die häufigeren autosomal-rezessiv vererbten, letalen Chondrodysplasien. Diese werden derzeit eingeteilt in SRPS I (Saldino-Noonan [MIM 269860]), SRPS II (Majewski [MIM 263520]), SRPS III (Verma-Naumhoff [MIM 263510]) und SRPS IV (Beemer [MIM 269860]). Pathognomonisch umfasst die Gruppe der SRPS die Kombination aus einer Wachstumsstörung auf Grundlage einer Skelettdysplasie mit Polydaktylien und engem Thorax. Weitere je nach Typ fakultative Fehlbildungen und Anomalien, wie urogenitale Fehlbildungen, insbesondere polyzystische Nieren, Herzfehler, Gehirnfehlbildungen, faziale Auffälligkeiten, wie Lippen-Kiefer-Gaumenspalten, und Hydrops fetalis können Bestandteil des Spektrums sein. Die Asphyxierende Thoraxdysplasie (ATD [MIM 208500]) und die Ellis-van Crefeld Syndrome (EVC [MIM 225500]) zeigen phänotypische Ähnlichkeiten zu den SRPSs, ebenso wie die Cranioektodermale Dysplasie (CED, Sensenbrenner Syndrom [MIM 218330]) bei der auch Haar, Zahn und Retina Anomalien beobachtet werden.
Als verursachender genetischer Defekt wurden zuerst bei einigen Patienten mit SRPS Typ III und ATD Mutationen in den ciliären Genen IFT80 und DYNC2H1 identifiziert. Mutationen im IFT122- und WDR35-Gen konnten als Ursache für Cranioektodermale Dysplasie und eine nicht näher klassifizierte Form des SRPS nachgewiesen werden. Alle Genprodukte sind hierbei am intraflagellären Transport (IFT) des primären Ciliums involviert.
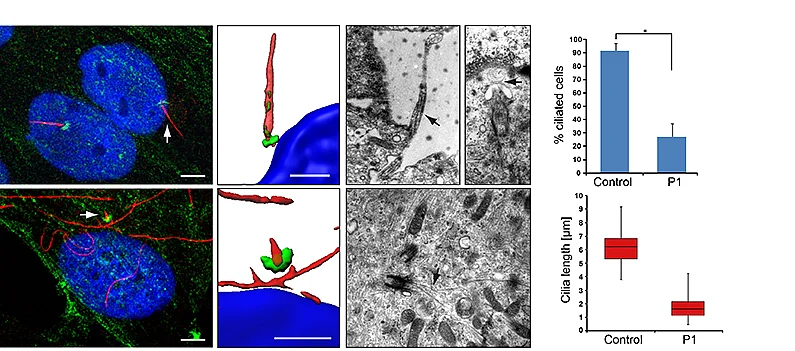
Zuletzt konnte die Arbeitsgruppe Mutationen im NEK1 Gen als Ursache für das SPRS II nachweisen (Thiel et al., 2011). Hierbei konnten wir nachweisen, dass Mutationen im humanen NEK1-Gen zu einer verminderten Ausprägung und zu einer morphologischen Veränderung des primären Ciliums durch einen Arrest in der Cilienbildung im Anfangsstadium führt. Das primäre Cilium spielt eine wichtige Rolle während der Entwicklung, ist an der Perzeption von mechanischen und chemischen Signalen beteiligt und koordiniert eine Reihe von Signaltransduktionswegen der Skelettentwicklung. Somit konnten wir hier den Zusammenhang zwischen der genetischen Ursache, dem zu Grunde liegenden molekularen Defekt und dem klinischen Krankheitsbild aufdecken.

